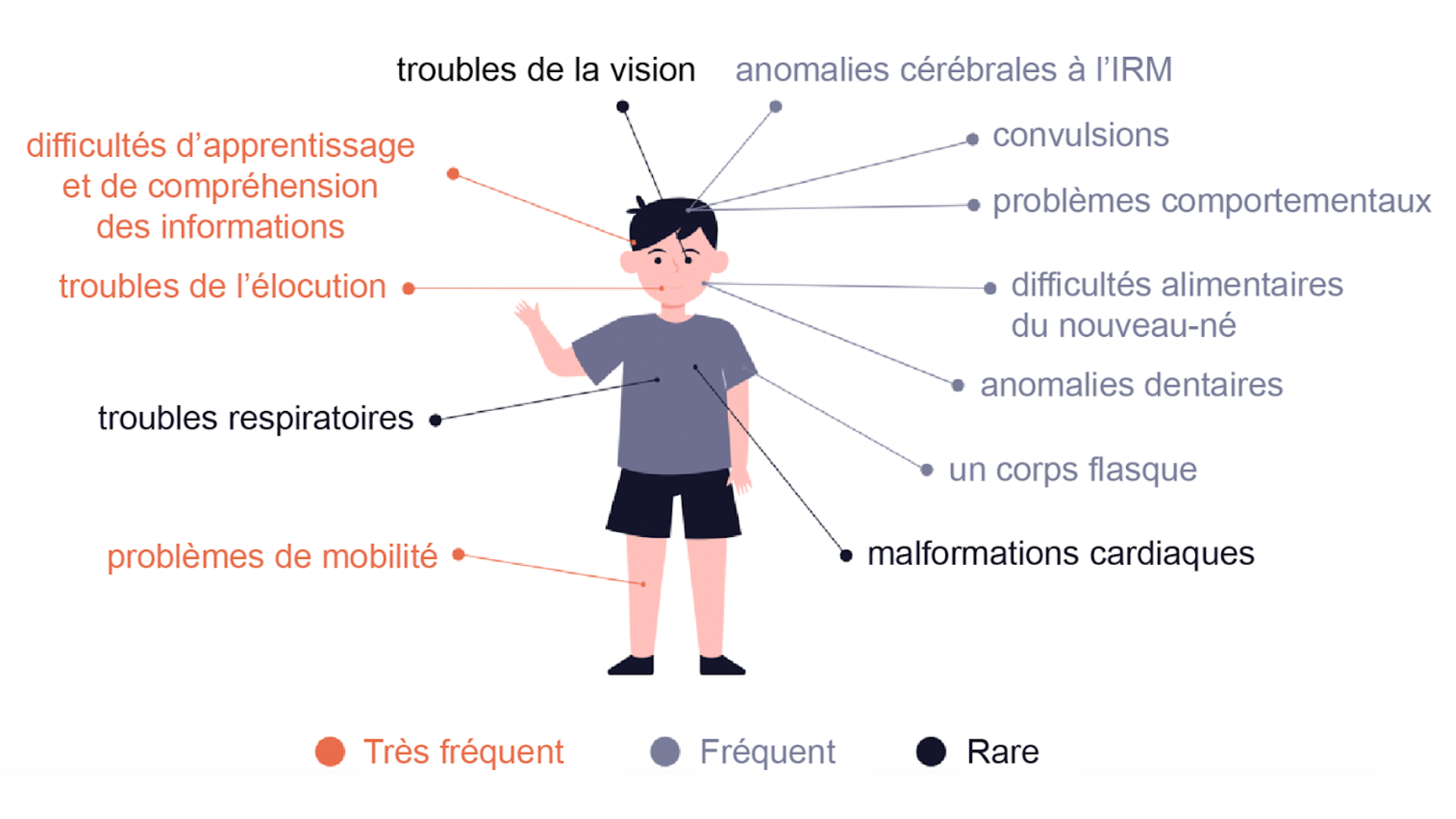
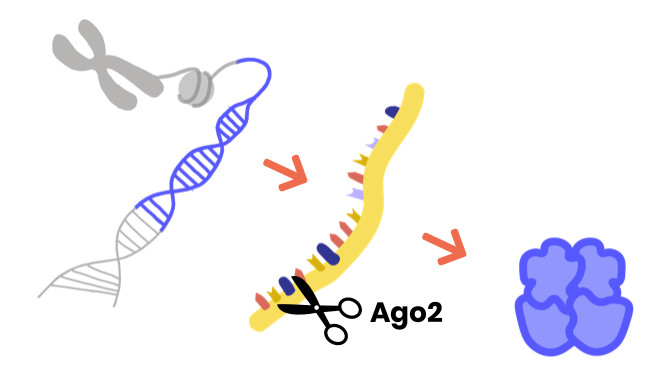
La modification du gène AGO1/2 est survenue naturellement et n’a pas été causée par le mode de vie, le régime alimentaire ou d’autres facteurs environnementaux. Le changement s’est produit de manière aléatoire soit dans le sperme ou l’ovule d’un parent, soit au début du développement de l’embryon après la combinaison de l’ovule et du sperme. Si vous êtes le parent d’une personne affectée, sachez que VOUS N’AVEZ RIEN FAIT DE MAL !
Nous avons tous deux copies de chaque gène Argonaute. Une mutation dans une seule des deux copies de l’AGO1/2 peut provoquer la maladie (« autosomique dominante »).
Chez la plupart des personnes diagnostiquées jusqu’à présent, aucun des parents ne présente la même modification génétique de l’AGO1/2. Les changements génétiques qui ne surviennent pas chez l’un ou l’autre des parents sont appelés mutations « de novo ».
Si le changement s’est produit « de novo », la probabilité que cela se reproduise pour un autre enfant est très faible. Dans de rares cas, plusieurs des ovules ou des spermatozoïdes sont porteurs de la même modification génétique (osaicisme gonadique), puis peuvent être transmis à un enfant. Si vous le souhaitez, vous pouvez effectuer un test de dépistage d’une modification du gène AGO1/AGO2 pendant la grossesse par amniocentèse. Demandez conseil à un généticien clinique.