Argonaute症候群は、Argonaute遺伝子AGO1及びAGO2の変異を原因とする疾患群です。
AGO2遺伝子の変異に関連する疾患は、Lessel-KreienkampまたはLeskres症候群とも呼ばれます。
Argonaute症候群は、Argonaute遺伝子AGO1及びAGO2の変異を原因とする疾患群です。
AGO2遺伝子の変異に関連する疾患は、Lessel-KreienkampまたはLeskres症候群とも呼ばれます。
症状は通常、乳児期又は幼児期に発現し、重症度は大きく異なり、広範囲に及びます(Lessel et al. 2020, OMIM, Schalk et al. 2021年)。
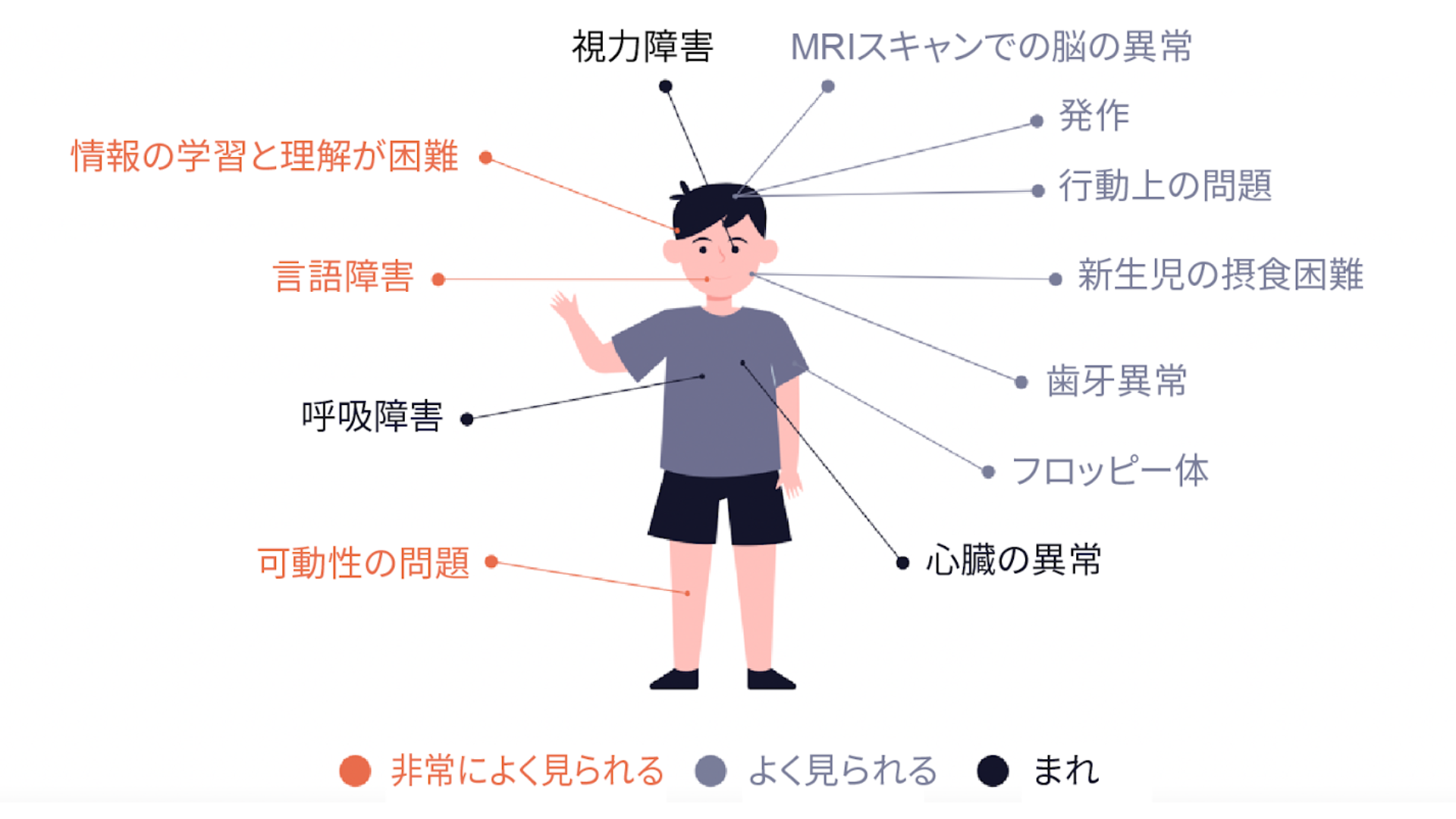
非常によく見られる
発話障害:発話の遅延から非言語
運動の問題:軽度の歩行遅延から車椅子生活へ
情報の学習と理解が困難:軽度から重度の知的障害
よく見られる
フロッピー体
発作、 WestおよびLennox-Gastau症候群
新生児の摂食困難
不安、多動性、自閉症または攻撃的行動などの行動上の問題
MRIスキャンでの脳の異常
歯牙異常
睡眠不足
低頻度
心臓の異常
視力障害
呼吸障害、出生時の救急、睡眠時無呼吸
病態は非常に新しいため、予後についてまだあまり分かっていません。患者レジストリを設定することで、予後を改善したいと考えています。
非常にまれです。世界中で150~200人の患者が診断されている。患数は増加しており、今後数年のうちにより多くの人が診断されることが予想されます。
最近この症候群が発見されたこと,および遺伝子パネルに遺伝子が現在含まれていないという事実を考慮すると,多くの患者は診断されていない可能性が高いです。科学者らは、遺伝子の長さと遺伝子配列に基づき、また遺伝子のコピー1つで十分な変異が存在するため、小児10万人に11人も罹患する可能性があると予測しました。ただし、これらの子供の一部は生まれてこない可能性があります。それでもなお、診断されていない子供がたくさんいる可能性があります!
Argonaute症候群は、全ゲノム(すべての遺伝物質)または全エクソームシーケンシング(タンパク質を作るよう指示されたゲノムの一部のみ)の遺伝子検査によって診断されます。
お子様が遺伝子検査を受け、報告書をどのように解釈してよいかわからない場合は、ClinGenによるこの101遺伝子ウェビナー(5-30分、他の言語にはキャプションと翻訳を使用):さまざまな遺伝子変化(挿入、欠失、ミスセンス、ナンセンス、重複、フレームシフト、c.1091T>C、p.Met364Thr)および一般的な用語(病原性、ヘテロボー、不確定バリアント)について学習します。
Argonauteタンパク質は、その発現をサイレンシングすることによって他の遺伝子を調節します。
遺伝子はDNAで構成され、体内で多くの異なる役割を果たすタンパク質を作るための細胞への指示を含んでいます。まず、遺伝子をmRNAに転写してから、タンパク質に翻訳します。アルゴナウトタンパク質は、RNA誘導サイレンシング複合体(RISC)の主要なメンバーです。RISCはmRNAを破壊することで遺伝子からタンパク質への過程を停止させることができ、遺伝子発現は停止されます。AGO1/2はmRNA標的を認識する上で重要な役割を果たし、AGO2はmRNAを直接切断する(「スライス」)こともあります。
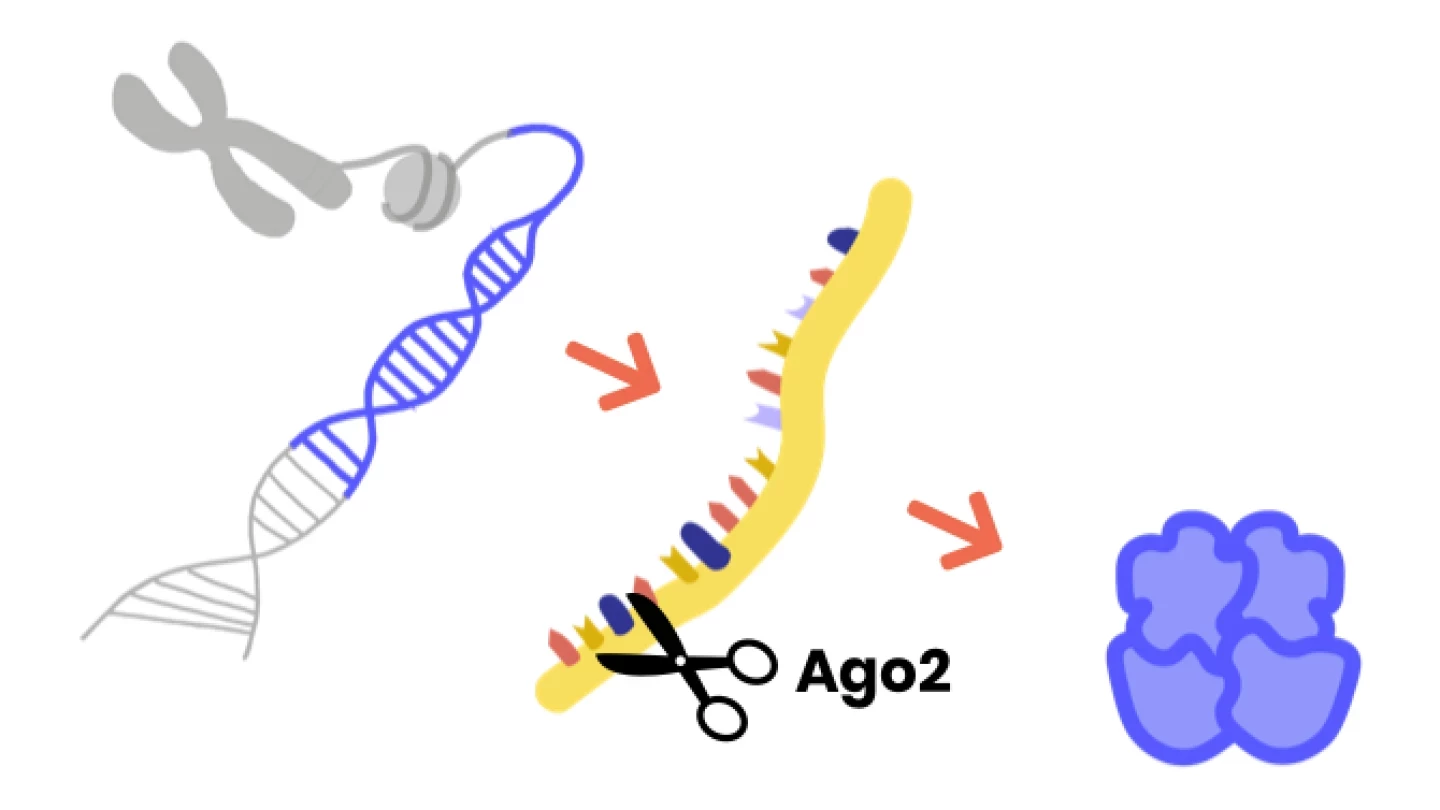
この1分間のビデオは、それを簡単な言葉で説明している。
アルゴノートタンパク質と結合するマイクロRNAの発見は、2024年にノーベル賞を受賞した。ノーベル賞受賞記者会見では、ビクター・アンブロスがアルゴノート症候群とマイクロRNAの関連性について考察している。
DNA、遺伝子、タンパク質の詳細については、ClinGenのこの101遺伝子ウェビナーをご覧ください(2~5分)。
Argonauteタンパク質とRNA誘導遺伝子調節の詳細については、Meister教授の著書「Meister Nature RevGenetics 2013」(アクセスできない場合はEメール)をご覧ください。
AGO1/2が変異している場合,遺伝子サイレンシングによる遺伝子発現の調節が障害されます。
正確な疾患を引き起こす機序については、まだ研究中です。
正確な発症メカニズムは現在も研究されている。
AGO1/2の変異により、AGO1/2の一部であるRNA誘導サイレンシング複合体(RISC)のサイレンシング活性が阻害され、遺伝子発現の調節が阻害される。Lesselらは、RISC複合体の形成不良および標的mRNAへの結合の増強を観察しました。患者由来皮膚細胞では、約20'000個中700~1'500個の遺伝子(4~8%)がアップレギュレートまたはダウンレギュレートされました(Lessel et al. 2020年)。AGO1/2は、全身、特に脳の発達における他の遺伝子の調節において重要な役割を果たします。
科学者らは現在、一部の遺伝的変化は、機能獲得(GOF)および機能喪失(LOF)の他の変化を伴うタンパク質につながると考えています。いずれの場合も,遺伝子調節に欠陥があります。疾患の機序は研究段階にあります。
ヒトには、AGO1からAGOAGO4と呼ばれる4つのアルゴネイトタンパク質があります。AGO3の欠失も神経発達障害と関連しています(Tokitaら 2015年)。
あなたの家族が別のアルゴネート遺伝子の変異に罹患している場合は、私的なFacebookサポートグループにもご参加いただけることをお知りおきください。
残念なことに、治療法や直接的な治療法はありません。
理学療法、言語療法、作業療法、または発作薬は、症状を管理し、お子さんの発達をサポートするのに役立ちます。担当の小児科医は、発達を監視し、確実に支援が与えられるようにケアを監督することができます。てんかんを専門とする神経内科医を受診する親もいます。
お子さんを過小評価せず、その発達をサポートすることを決して止めないでください!Lesel教授は、「即時に進展が見られなくても、サポート、サポート、サポートを奨励しています」。
お子さまが診断された場合は、プライベートのFacebookサポートグループにご連絡ください!この症候群が希少なことを考えると、現在、他の罹患家族が医師よりも病気について多くを知っている可能性があります。
希望があります。遺伝子治療は過去10年間で大きな進歩を遂げ、最終的に患者のベッドサイドに到着しました。
筋消耗性疾患である脊髄性筋萎縮症(SMA)に対する、SpinrazaとZolgensmaの2つの遺伝子治療が2016年と2019年に、承認されました。
神経発達疾患であるAngelman症候群に対するRNA遺伝子治療の臨床試験が開始されました。
転用薬は希少疾患Progeriaに対して承認されており、その他は治験(PMM2-CDG、SLC6A1)に近接している/治験中である。
希少疾患コミュニティは、インキュベーターとして作用し、医薬品開発の初期段階のリスクを減らすことで、可能性の限界を押し広げるのに役立ちます。
疾病負荷および患者の人口統計学的特性に関する情報の収集
患者の組織と動物モデルを利用可能にする、
転帰指標の定義
疾患機序を明らかにし、薬剤候補の概念実証を確立するための研究に資金提供する。
科学アドバイザーと皆様のご支援とともに、私たちは治療と治癒に向けて絶え間なく努力していきます。
まず、どの変異がタンパク質(GOF)の機能獲得につながるのか、どの変異が機能喪失(LOF)につながるのかを理解する必要があります。なぜなら、治療戦略は、GOFの変異タンパク質を止め、LOFの健康なタンパク質を増加するという、互いに反対であるからです。
当社は、治療を見つけるために以下の治療戦略を追求することを目指しています。
オフターゲット効果のスクリーニングですでに承認または開発された医薬品から開始するAGO1/2の産生または活性を低下させる/増加させる低分子医薬品。
スプライシングアンチセンスオリゴヌクレオチド(ASO)又は調節エレメントASOを用いて行う、AGO1/2の産生を停止/上方制御するためのRNA療法
AGO1/2の機能的コピーを送達するDNA療法(AAV媒介遺伝子療法-LOFのみ)又は変異を修正する(CRISPR塩基編集)。CRISPRベース編集は、ヒト療法から何年も経過しています。
本ウェブサイト上の情報は、教育および情報提供のみを目的としており、医学的な助言を提供するものではありません。本ウェブサイトのいかなる資料も、専門的な医学的助言、診断、または治療に代わるものではありません。