Argonaute syndromes are a group of rare disorders associated with disease-causing changes in the Argonaute genes AGO1 (AGO1-related syndrome) and AGO2 (Lessel-Kreienkamp or Leskres syndrome).
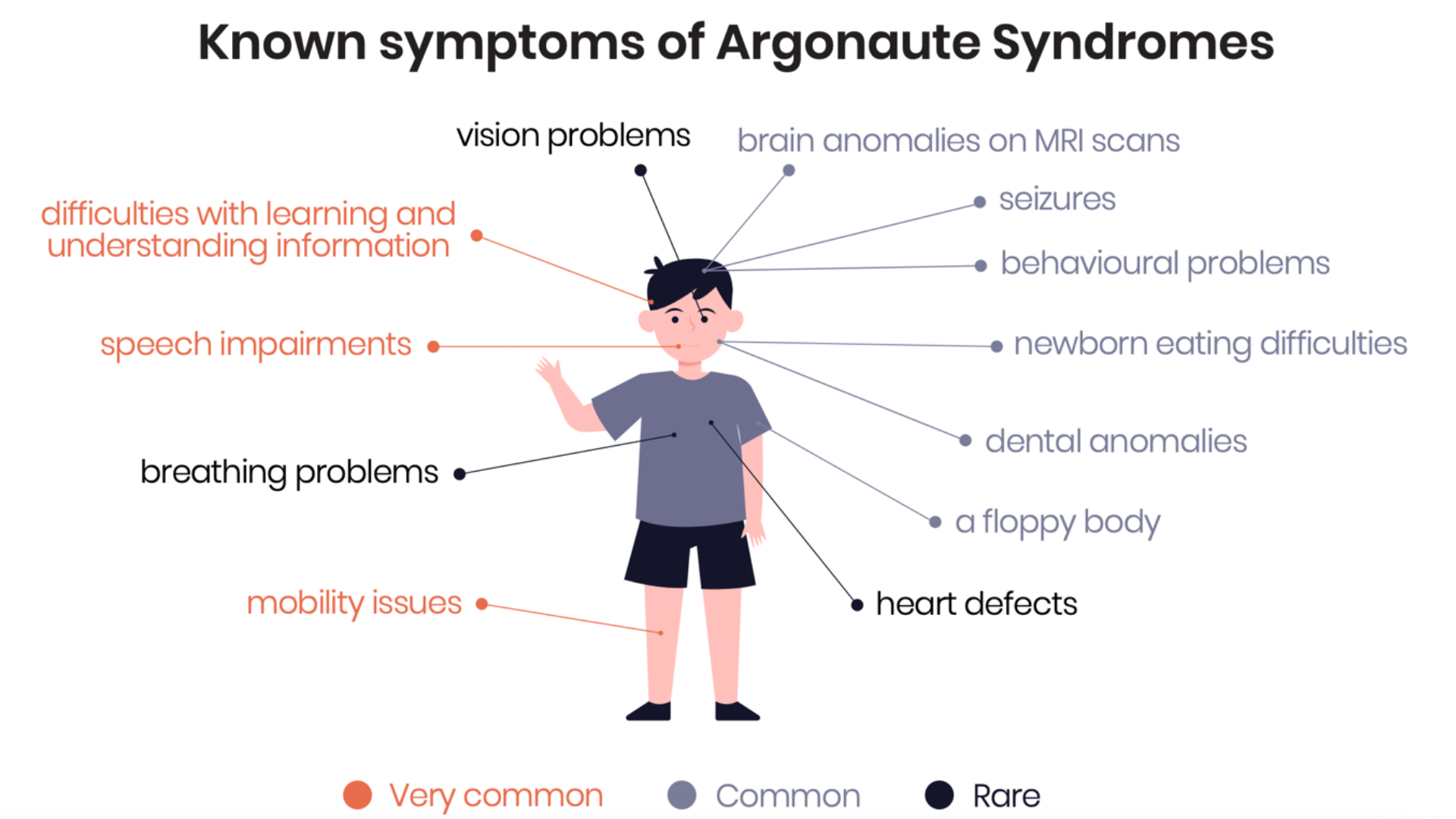
Argonaute syndromes are rare neurodevelopmental disorders characterized by delayed motor development, seizures, problems speaking and understanding, and cognitive impairment.
They are associated with disease-causing changes in the Argonaute genes, specifically Argonaute1 (AGO1) and Argonaute2 (AGO2).
The AGO1/2 gene change occurred randomly and was not caused by lifestyle, diet, or other environmental factors. It happened either in the sperm or egg of a parent, or during early development of the embryo. If your child is affected, please know that YOU DID NOTHING WRONG!
We all have two copies of AGO1 and AGO2. A mutation in a single copy can cause the disorder ("autosomal dominant").
In most people diagnosed so far, neither parent has the same genetic change in AGO1/2. Such novel genetic changes are called “de novo” mutations and it means that the likelihood of another child being affected is low (1-2%). If neither parent has the mutation in any of their cells, the risk is ~0.1% (1 in 1'000).
There is a catch. A recent study by Kay et al. (J Med Genet 2023) found that in ~10% of families, the egg or sperm cells of one of the parents carried the same genetic change (gonadal mosaicism), which means that it can be passed to another child and that risk is up to 50%. If you wish, you can test for an AGO1/AGO2 gene change during pregnancy via amniocentesis. Ask a clinical geneticist for advice.
mobility issues: from mildly delayed walking to wheelchair-bound
difficulties with learning and understanding information: from mild to severe intellectual disability
Common
a floppy body
seizures, incl. West and Lennox-Gastaut syndromes
newborn eating difficulties
behavioral problems, e.g. anxiety, hyperactivity, autistic or aggressive behavior
dental anomalies
brain anomalies on MRI scans
poor sleep
Uncommon
heart defects
vision problems
breathing problems, esp. at birth, and sleep apnea
You can learn more about the latest knowledge on Argonaute Syndromes in the Natural History Study presentation at the Argonaute Syndromes Conference 2025.
As the condition is so new, we still don't know enough about its diversity and prognosis - we want to help change that! If your child was diagnosed with an AGO1 or AGO2 mutation anywhere in the world, please participate in the Natural History Study led by Prof. Lessel.
They are very rare. Worldwide, 150-200 patients have been diagnosed so far. The number is growingand we expect many more individuals will be diagnosed in the coming years.
Given the recent discovery of the syndrome and the fact that the gene is not included in many gene panels, many patients are likely undiagnosed. Scientists predicted that as many as 11 in 100´000 children may be affected, based on the gene length & sequence, and because a mutation in one copy of the gene is enough. It is possible, however, that some of these children are never born. Still, this means that there could be many undiagnosed children!
Argonaute syndromes are diagnosed via a genetic test: either a gene panel (small subset of genes tested), whole genome (all your genetic material) or whole exome sequencing (only the part of your genome with instructions to make proteins).
If your child had genetic testing done and you do not know how to interpret the genetics report, have a look at the brochure "Understanding your child's genetics report", created by genetic counselling student Hannah Sandler, for Argonaute syndrome parents: web version, print version.
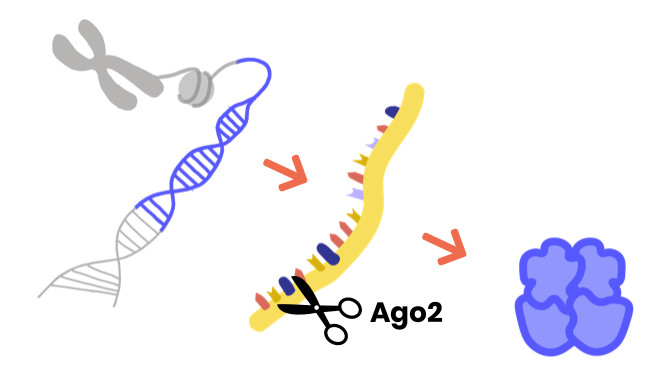
The Argonaute proteins regulate other genes by silencing their expression.
A gene is made up of DNA and contains instructions for your cells to make proteins, which play many different roles in your body. First, the gene is transcribed into mRNA, and then it is translated into a protein. Argonaute proteins are key members of the RNA-induced silencing complex (RISC). RISC can stop the journey from gene to protein by destroying the mRNA: gene expression is silenced. AGO1/2 plays a crucial role in recognizing the mRNA target, and AGO2 may also directly cut ("slice") mRNAs.
The discovery of microRNA, which binds to Argonaute proteins, was awarded the Nobel Prize in 2024. In the Nobel Prize News Conference, Victor Ambros reflects on the relevance of microRNA to Argonaute Syndromes.
Learn more about DNA, genes and proteins in this 101 Genetics webinar from ClinGen (minutes 2-5).
Learn more about Argonaute proteins and RNA-guided gene regulation in Prof. Gunter Meister's excellent review Meister Nature Rev Genetics 2013 (email us if you can't access it).
Regulation of gene expression via gene silencing is defective
The disease-causing mechanisms are still under study
Because of the mutation in AGO1/2, the silencing activity of the RNA-induced silencing complex (RISC), which AGO1/2 is a part of, and the regulation of gene expression are disturbed: Lessel et al. observed faulty formation of the RISC complex and enhanced binding to target mRNAs. 700-1'500 genes out of ~20'000 (4-8%) were up- or downregulated in patient-derived skin cells (Lessel et al. 2020). AGO1/2 play a crucial role in the regulation of other genes all over the body, and in particular in thedevelopment of the brain.
Scientists currently believe that some genetic changes lead to a protein with a gain of function (GOF) and other changes to a loss of function (LOF). In either case, gene regulation is defective. The mechanism of disease is under study.
To learn more about gene variants (missense, deletion), read our guide to understanding genetics reports.
Humans have 4 Argonaute proteins, called AGO1 to AGO4. Deletions in AGO3 have also been associated with a neurodevelopmental disorder (Tokita et al. 2015).
If your family is affected by a mutation in another Argonaute gene, know that you are welcome to join our private Facebook support group too!
In 1998, Bohmert et al. named the gene after Argonauta argo, a shell octopus, because the Arabidopsis plant lacking AGO1 looks like a little squid.
Our mascot is called 'Archie', and is modeled after a clay figure designed by Alex, a child affected by an AGO1 mutation.
There is unfortunately no cure and no direct treatment... yet.
Physical, speech, and occupational therapies or seizure medication can help to manage symptoms and support your child's development. Your pediatrician can oversee care to monitor development and ensure help is given. Several parents also see a neurologist specializing in epilepsy.
Do not underestimate your child and never stop supporting their development! Prof. Lessel encourages "support, support, support even if you don’t see immediate progress".
If your child was diagnosed, please reach out to us and join our private Facebook support group! Given the rarity of the syndrome, other affected families may currently know more about the disease than doctors.
There is hope. Genetic therapies have made tremendous progress in the last decade and have finally come to the patient's bedside.
Two gene therapy treatments, Spinraza and Zolgensma, for the muscle-wasting disease spinal muscular atrophy (SMA) were approved in 2016 and 2019,
RNA-based gene therapy for Angelman syndrome, a neurodevelopmental condition, has started clinical trials,
A repurposed drug was approved for the rare disease Progeria and others are close to/in a trial (PMM2-CDG, SLC6A1).
Rare disease communities help to push the boundaries of the possible by acting as incubators and by de-risking the early stages of drug development by:
collecting information on disease burden and patient demographics,
making patient tissue and animal models easily available,
defining outcome measures,
funding research to elucidate disease mechanisms and establishing proof of concepts for drug candidates.
Together with our scientific advisors and your support, we will work incessantly towards treatments and a cure.
First, we need to understand which mutations lead to a gain of function of the protein (GOF) and which ones to a loss of function (LOF), because therapeutic strategies are the opposite of each other: stop the mutated protein for GOF, and increase the healthy one for LOF.
We aim to pursue the following therapeutic strategies to find treatments:
Small molecule drugsto decrease/increase production or activity of AGO1/2, starting with screening already approved or developed drugs for off-target effects
RNA therapyto stop/upregulate the production of AGO1/2 with a splicing antisense oligonucleotide (ASO) or regulatory element ASO
DNA therapy to deliver a functional copy of AGO1/2 (AAV-mediated gene therapy – LOF only) or correct the mutation (CRISPR base editing). CRISPR base editing is many years from human therapies.
The information on this website is for educational and informational purposes only and does not provide medical advice. No material on this website is intended to be a substitute for professional medical advice, diagnosis or treatment.