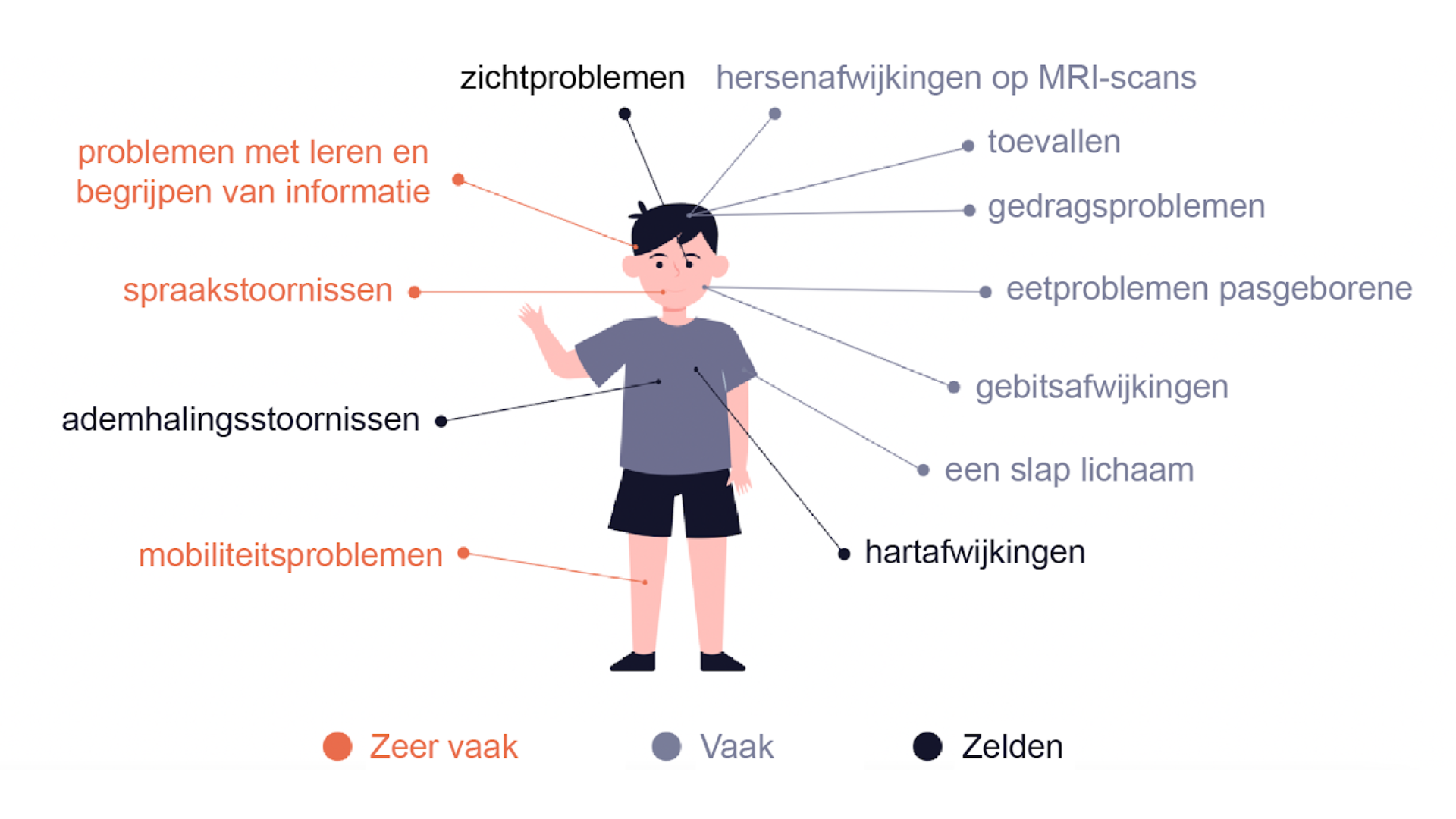
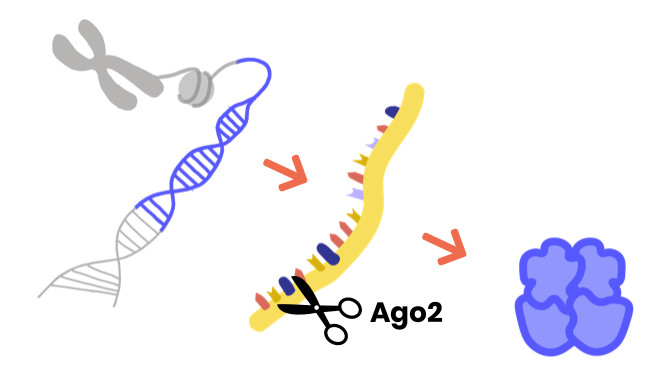
Argonaute-syndromen zijn zeldzame neurologische ontwikkelingsstoornissen die gekenmerkt worden door vertraagde motorische ontwikkeling, epileptische aanvallen, problemen met praten en begrijpen en cognitieve stoornissen.
Ze worden geassocieerd met ziekteveroorzakende veranderingen in de Argonaute-genen, met name Argonaute1 (AGO1) en Argonaute2 (AGO2).
Prof. Piton beschreef het AGO1-gerelateerd syndroom in 2021. Prof. Lessel en Prof. Kreienkamp ontdekten het AGO2-gerelateerde syndroom en beschreven het voor het eerst in november 2020. Dit laatste werd begin 2021 het “Lessel-Kreienkamp-syndroom” (OMIM) genoemd.