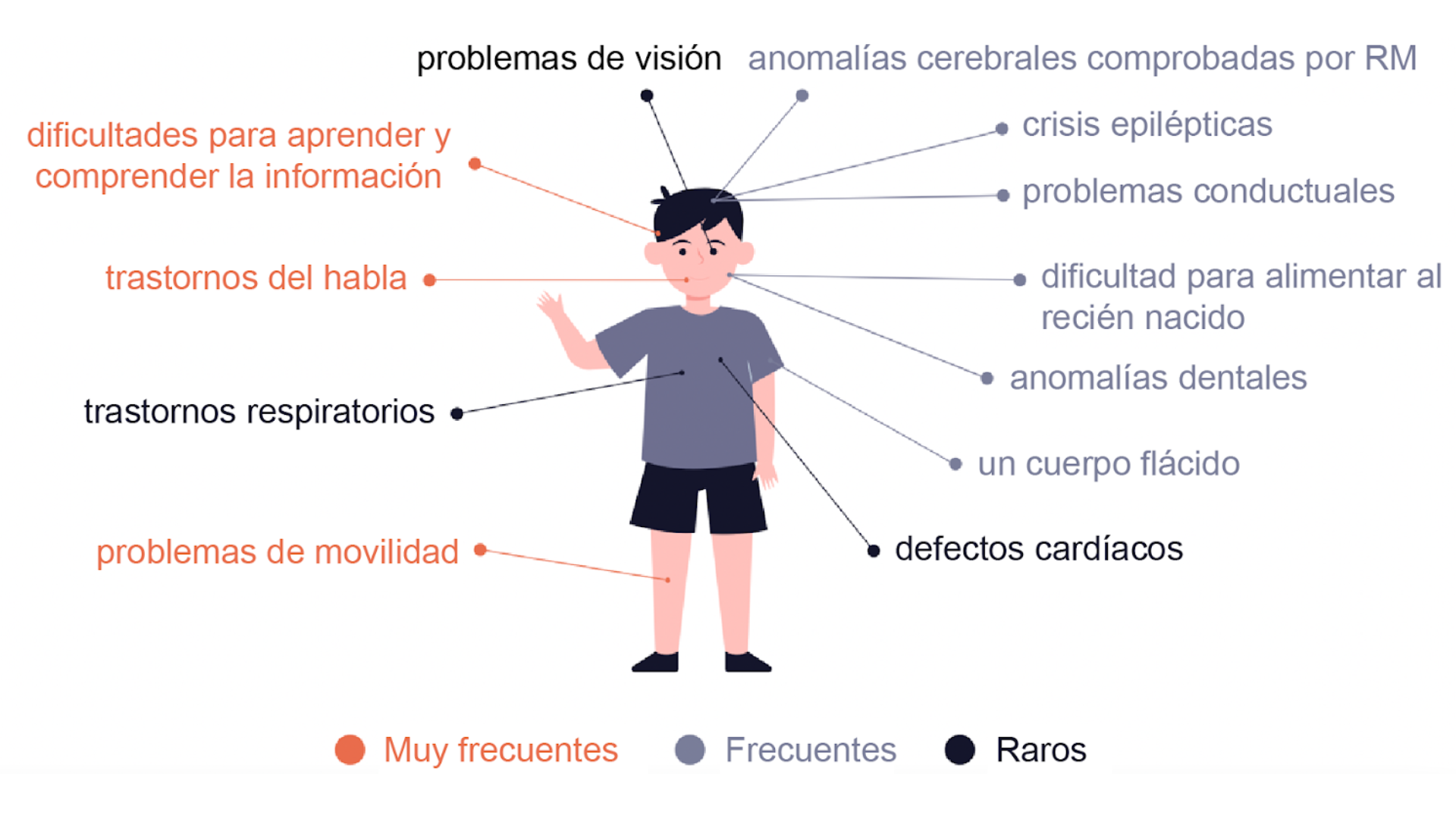
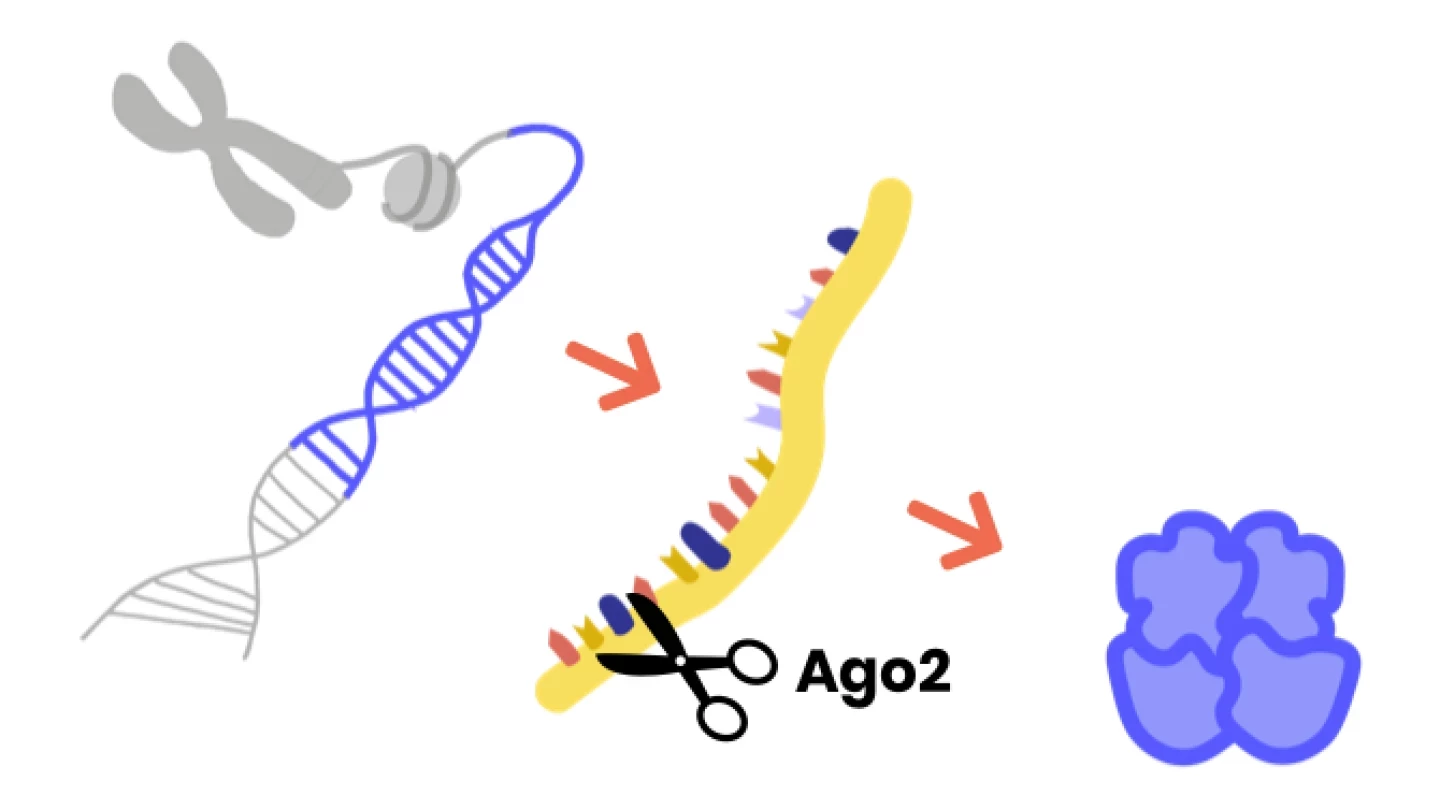
Los síndromes del Argonauta son trastornos raros del neurodesarrollo caracterizados por un desarrollo motor tardío, convulsiones, problemas para hablar y comprender y deterioro cognitivo.
Están asociados con cambios patológicos en los genes de las proteínas Argonauta, específicamente Argonauta1 (AGO1) y Argonauta2 (AGO2).
El profesor Piton describió el síndrome relacionado con mutaciones en AGO1 en el 2021. Tanto el prof. Lessel como el prof. Kreienkamp descubrieron el síndrome relacionado con mutaciones en AGO2 y lo describieron por primera vez en noviembre de 2020. Este segundo síndrome se denominó “síndrome de Lessel-Kreienkamp” a principios de 2021 (OMIM).