Study title: Dysregulation of AGO2-miRNA dynamics underlies the AGO2-associated Lessel–Kreienkamp syndrome
New research by Profs. Lessel, Kreienkamp and MacRae helps us understand why AGO2 mutations lead to neurodevelopmental problems — it’s not just a broken gene, but a disrupted regulatory system. Understanding exactly how the mutation disrupts its normal function is an important step toward future therapies.
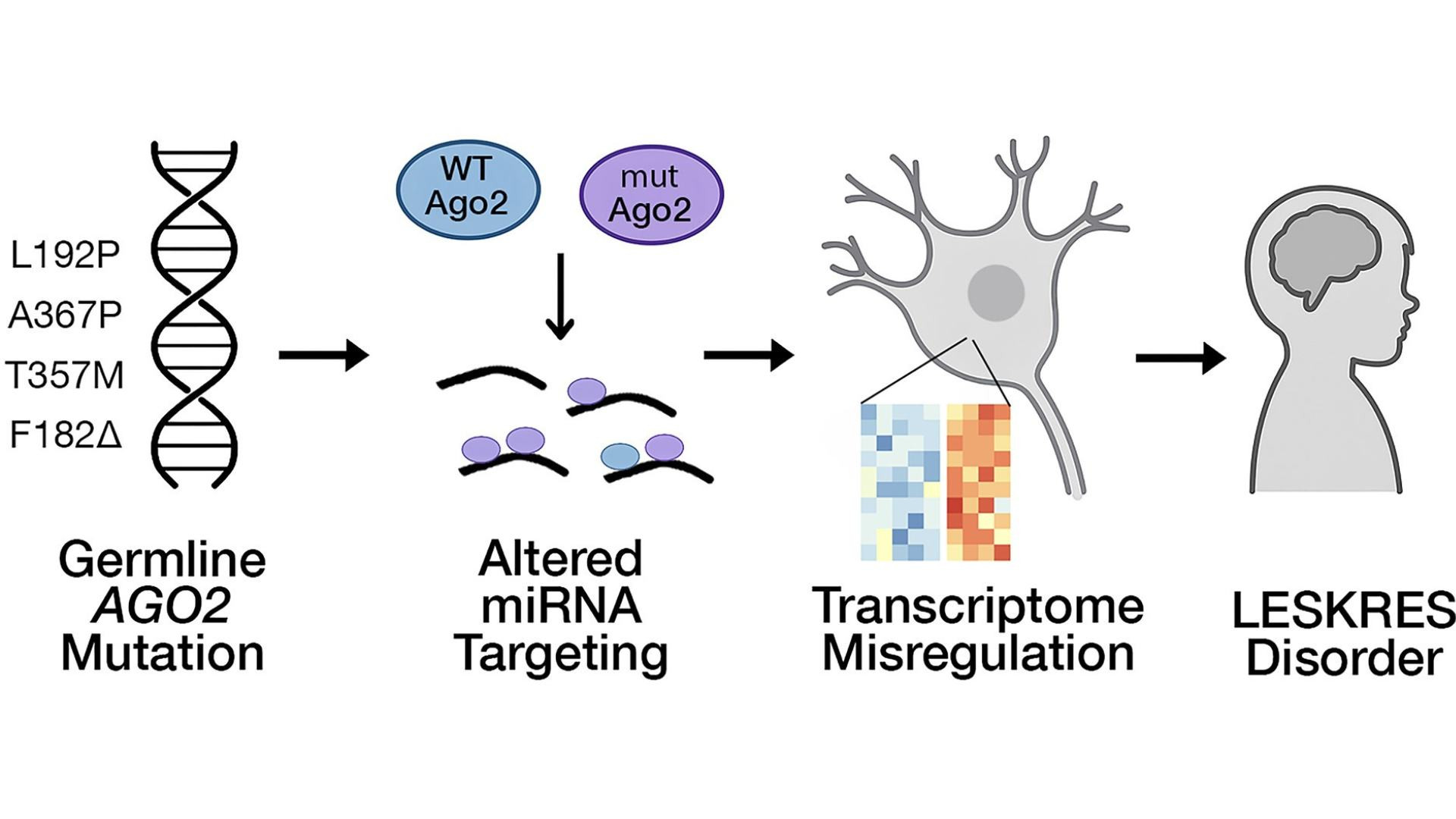
AGO2 (like AGO1) helps control how other genes are turned “on” or “off,” especially in the brain. When AGO2 doesn’t work properly, it can affect brain development, speech, movement, and learning in different ways.
Quick explainers
In vitro: Experiments done outside a living organism, for example in a cell culture in a dish.
Loss vs. gain of function:
A loss-of-function mutation makes a protein work less well or not at all.
A gain-of-function mutation causes the protein to do too much, act at the wrong time, or behave abnormally.
What did the researchers find?
Researchers studied how the AGO2 protein works together with tiny molecules called miRNAs. In healthy cells, AGO2 and miRNAs constantly attach and release from their targets in a balanced way — like a switch that keeps gene activity just right.
Binding and release: some mutations (L192P, A367P, T357M) caused the AGO2–microRNA complex to stay bound to target RNAs much longer than normal (i.e., slower release), and in the case of L192P and A367P, the association (binding) was also slower.
Increased “mis-targeting” in vitro: certain AGO2 mutants (L192P, A367P, T357M, F182del) showed a higher tendency to bind to non-ideal/mismatched targets — meaning they were less selective than normal.
Effects in mice: Mice engineered to carry the L192P variant (but not the G733R variant) showed marked changes: reduced offspring, changed gene expression profiles (transcriptomes) in the cortex, and over-repression of microRNA target genes.
With AGO2 mutations some RNAs that should be repressed may not be, or repression may become less precise. This results in widespread “noise” in gene activity, especially in brain cells.
The authors propose that these patient-linked AGO2 mutations result in a gain-of-function type problem (not simply loss of function).
Why does this matter?
This study reveals how specific AGO2 mutations associated with Lessel–Kreienkamp syndrome disrupt the delicate partnership between AGO2 and microRNAs. Instead of destroying AGO2’s function, many mutations make AGO2 behave abnormally: it binds too long, releases too slowly, becomes less selective, and loads microRNAs differently. These changes disturb gene regulation in neurons and lead to measurable effects in mice.
Importantly, the study shows that several of these mutations act through a gain-of-function mechanism — meaning the protein is present but behaving in an abnormal or excessive way. This distinction matters greatly for future therapy development:
Loss-of-function disorders typically require adding back a working version of the protein (e.g., gene replacement).
Gain-of-function disorders often require approaches that reduce, silence, or remove the harmful mutant protein, ideally while keeping the healthy copy active.
Understanding which mechanism applies is essential because it directly guides which therapeutic strategies are likely to help — and which might unintentionally worsen the problem.
While there’s no direct treatment yet, this research brings scientists closer to understanding the root cause and helps shape the scientific direction toward targeted, mechanism-appropriate therapies.
References
For more details, you can read the full paper for free here.
Liu et al., Dysregulation of AGO2-miRNA dynamics underlies the AGO2-associated Lessel–Kreienkamp syndrome. Nucleic Acids Research. 2025; 53(19):gkaf1002